Are you ready for Brexit? Do you have a Brexit contingency plan?
You may need either an EU/EC European authorized representative based in EU-27 countries or a UK authorised representative (so-called "UK Responsible Person") based in UK, or may even need both EU & UK representatives, depending on different brexit scenarios.
Register/Notify your MD-Medical Devices & IVD-In Vitro Diagnostic Medical
Devices with MHRA in UK & other EEA (EU/EFTA) authorities by world-leading
consultancy- Wellkang team based in both UK (England) & EU-27 (Ireland).
Wellkang team can help you under all Brexit scenarios!
Click here to get FREE Guide Now! |
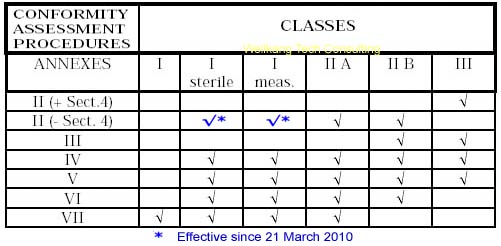
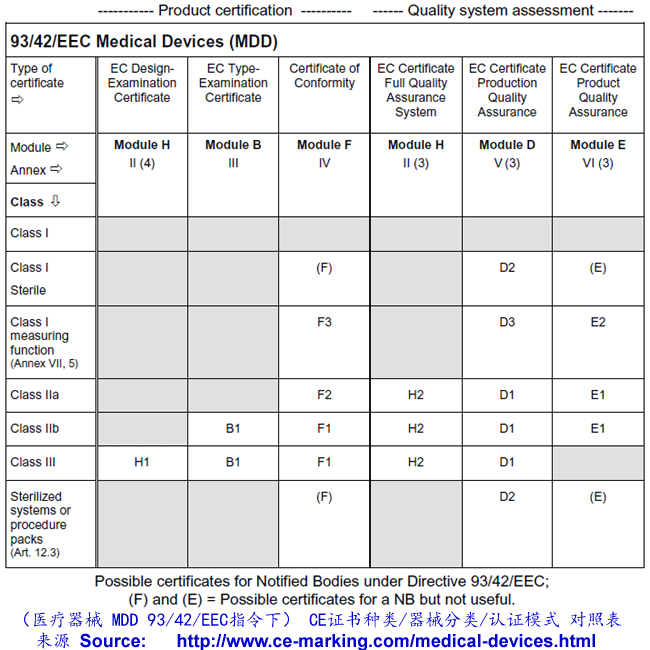
Steps for Class IIb medical devices compliance
- Classification: ensure the device is a Class IIb medical device.
- Choose Conformity Assessment Route: refer the flow chart below.
- Compile the Technical File.
- Obtain certification from a Notified Body
- Declaration of Conformity.
- Appoint an Authorised Representative. (Hold the Tech Files for inspection by the Competent Authority)
- Vigilance and Post Market Surveillance. (affix CE marking & market the products)
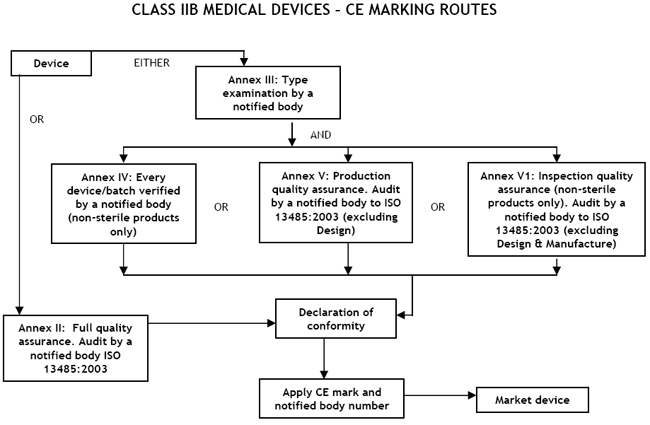
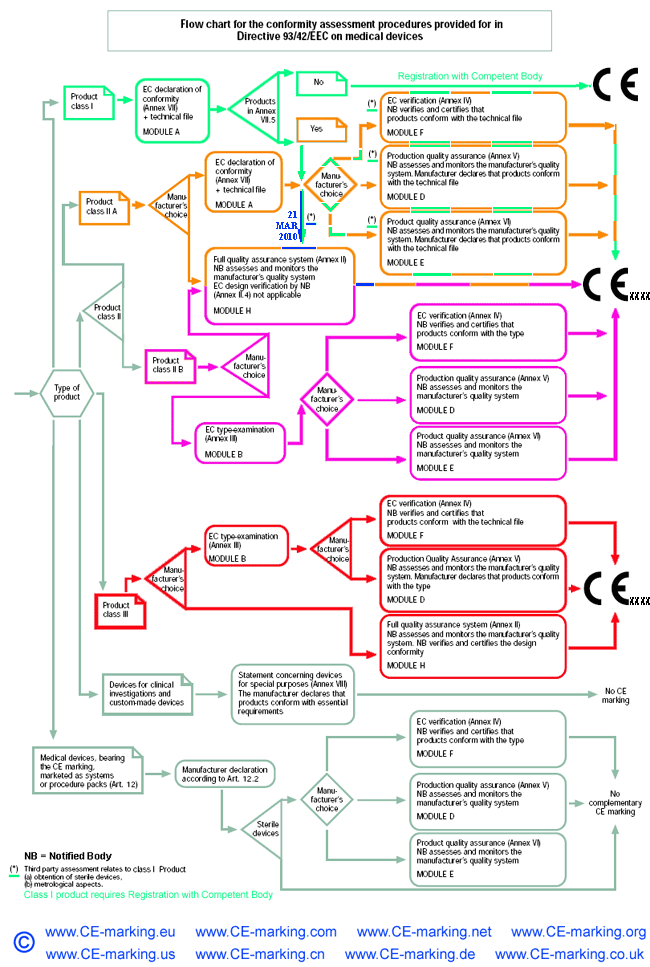
Class IIb Medical Devices: Conformity Assessment Routes
The conformity assessment routes for Class IIb Medical Devices
In the case of devices falling within Class IIb, other than devices
which are custom-made or intended for clinical investigations, the
manufacturer shall, in order to affix the CE marking, either:
- follow the procedure relating to the EC declaration of conformity
set out in Annex II (full quality assurance); in this case, point 4 of
Annex II is not applicable;
or
- follow the procedure relating to the EC type-examination set out in
Annex III, coupled with:
- (i) the procedure relating to the EC verification set out in Annex
IV;
or
- (ii) the procedure relating to the EC declaration of conformity set
out in Annex V (production quality assurance);
or
- (iii) the procedure relating to the EC declaration of conformity set
out in Annex VI (product quality assurance).
There are two routes:
- a Notified Body must carry out either an Annex II audit of the full
quality assurance system (ISO 13485:2003),
or
- a type-examination (Annex III) plus one
of the three options given here:
- Examination and testing of each product or homogenous batch of products (Annex IV);
or
- Audit of the production quality assurance system (Annex V:) ISO 13485:2003 (excluding
Design) or
- Audit of final inspection and testing (Annex VI:) ISO 13485:2003 (excluding Design &
Manufacture)
Once the manufacturer has received certification from the Notified Body he may CE mark his
products and place them on the market.

FAQ/Q&A: Questions and Answers about CE Marking of Medical Devices
About CE Marking:
| Home | About Us | Contact Us | Copyright & Disclaimer | Privacy | Log-in | Order Now | |