
Are you ready for Brexit? Do you have a Brexit contingency plan?
You may need either an EU/EC European authorized representative based in EU-27 countries or a UK authorised representative (so-called "UK Responsible Person") based in UK, or may even need both EU & UK representatives, depending on different brexit scenarios.
Register/Notify your MD-Medical Devices & IVD-In Vitro Diagnostic Medical
Devices with MHRA in UK & other EEA (EU/EFTA) authorities by world-leading
consultancy- Wellkang team based in both UK (England) & EU-27 (Ireland).
Wellkang team can help you under all Brexit scenarios!
Click here to get FREE Guide Now! |
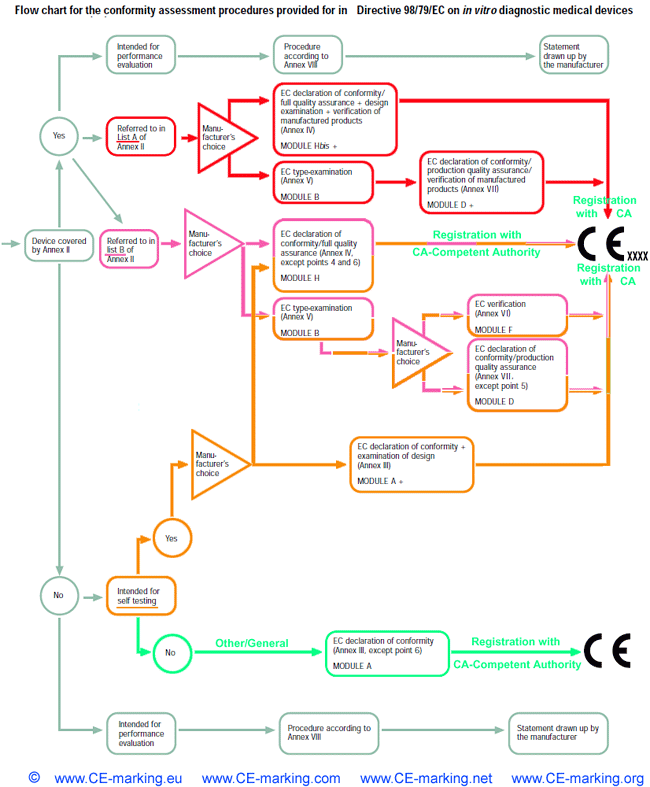
IVD-In Vitro Diagnostic Medical Devices: Conformity Assessment Routes
-
For all devices referred to in List A in Annex II
other than those intended for performance evaluation, the manufacturer shall, in order to
affix the CE marking either:
- (a) follow the procedure relating to the EC declaration of conformity
set out in Annex IV (full quality assurance),
or
- (b) follow the procedure relating to EC type-examination set out in
Annex V coupled with the procedure relating to the EC declaration
of conformity set out in Annex VII (production quality assurance).
-
For all devices referred to in List B in Annex II
other than those intended for performance evaluation, the manufacturer shall for the
purposes of affixing the CE marking, follow either:
- (a) the procedure relating to the EC declaration of conformity set out in
Annex IV (full quality assurance)
or
- (b) the procedure relating to EC type-examination set out in Annex V
couplet with:
(i) the procedure relating to EC verification set out in Annex VI,
or
(ii) the procedure relating to the EC declaration of conformity set
out in Annex VII (production quality assurance).
- Other than those covered by Annex II
- Self-testing
For all devices for self-testing other than those covered by Annex II and devices for performance evaluation, the manufacturer shall, prior to the drawing up the EC declaration of conformity in Annex III, fulfill the supplementary requirements set out in Annex III, point 6. Instead of
applying this procedure, the manufacturer may follow the procedure
referred to for products in List A or List B in Annex II.
- Other/General
For all devices other than those covered by Annex II and devices
for performance evaluation, the manufacturer shall, in order to affix the
CE marking, follow the procedure referred to in Annex III and draw up
the EC declaration of conformity required before placing the devices on
the market.
- In the case of devices for performance evaluation, the manufacturer shall follow the procedure referred to in Annex VIII and draw up the statement set out in that Annex before such devices are made available.
This provision does not affect national regulations relating to the ethical aspects of carrying out performance evaluation studies using tissues or substances of human origin.
Examples of Registration
Confirmation Letter issued by UK Competent Authority MHRA for In Vitro Diagnostic Medical Device (IVD)

FAQ/Q&A: Questions and Answers about CE Marking of Medical Devices
About CE Marking:
| Home | About Us | Contact Us | Copyright & Disclaimer | Privacy | Log-in | Order Now | |
